
Introduction
Contamination is the single most common technical reason drugs get pulled from the market. In FDA recall data covering 2015-2025, roughly 15.3% of all drug recalls were directly attributed to contamination issues spanning microbial contamination, particulate matter, and cross-contamination between products.
A separate ten-year analysis of FDA recall causes found impurities and contaminants behind roughly 37% of recalls, more than any other single category, including labeling and specification failures combined.

For API manufacturers, this isn’t an abstract compliance risk it’s the difference between a batch that ships and a batch that triggers a Form 483, a warning letter, or an import alert.
Contamination control has consequently moved from being a quality-department checklist item to a boardroom-level investment priority, with global spending across cleanrooms, isolators, environmental monitoring, and automation all growing at double-digit rates.
This article examines why contamination control matters, what modern facilities are doing about it, and how buyers and quality teams can evaluate whether a given manufacturing operation is actually equipped to prevent it.
Why Contamination Control Is Critical in API Manufacturing
Contamination control in pharmaceutical manufacturing means preventing any unwanted biological, particulate, or chemical substance from entering a product at any stage of production.
Industry guidance frames this not as a single checklist but as an interconnected system a Contamination Control Strategy (CCS) that spans facility design, material management, staff training, and quality monitoring, built on a foundation of scientific knowledge, quality risk management, and a genuinely quality-focused culture.
The stakes are well documented in enforcement data. A 21-year review of FDA enforcement reports (2004-2025) tracking microbial contamination in drugs found persistent citations for organisms such as Burkholderia cepacia complex and Aspergillus species in both sterile and non-sterile products, with contamination identity often left unspecified in enforcement records itself a red flag about the rigor of underlying root-cause investigations.
Separately, the FDA’s own FY2024 quality enforcement report identified contamination ranging from microbial issues to particulates as the single top defect category behind recalled products that year, even as total recalled products fell to a five-year low.
For API manufacturers specifically, the consequences of failure are severe and compounding batch rejection, customer disqualification, regulatory action (warning letters, import alerts), and in the worst cases patient harm.
As one industry analysis puts it, contamination control is not just about product quality; it is fundamentally about patient trust and safety, making it a core pillar of Good Manufacturing Practice (GMP) rather than an optional add-on.
Modern Facility Design Plays a Major Role in Contamination Prevention
Contamination prevention starts before a single batch is manufactured it starts with how the facility itself is designed. Industry guidance identifies several non-negotiable design elements for contamination-controlled API production:
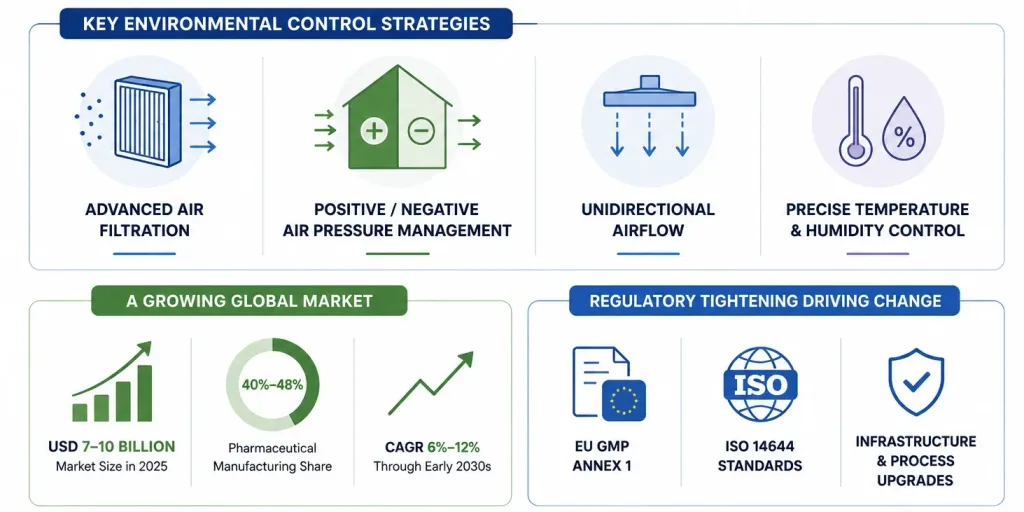
This is not a small market. The global cleanroom technology market the physical infrastructure underpinning most of this design philosophy was valued at roughly USD 7-10 billion in 2025 depending on scope and methodology, with most forecasts projecting it to more than double by the early 2030s at CAGRs in the 6-12% range.
Pharmaceutical manufacturing consistently represents the largest end-use segment, accounting for somewhere between 40% and 48% of total cleanroom technology demand depending on the market research source.
Regulatory tightening is a direct driver revisions to EU GMP Annex 1 and ISO 14644 cleanroom classification standards are pushing manufacturers worldwide to upgrade infrastructure, monitoring systems, and sterilization workflows rather than treating existing installations as permanently compliant.
Modular and prefabricated cleanroom construction is one of the fastest-growing segments within this market, in some analyses cutting installation time by up to 60% compared with traditional stick-built cleanrooms a meaningful advantage for API manufacturers needing to scale capacity quickly in response to demand shifts like the “China Plus One” sourcing trend.
How Automation Reduces Human Contamination Risks
Human presence remains one of the largest variable sources of contamination risk in any API facility, skin flakes, respiratory droplets, clothing fibers, and simple procedural error all introduce risk that no amount of gowning fully eliminates. Automation is the industry’s primary structural response.
Transfer Systems
Automated material and component transfer systems including rapid transfer ports, pass-throughs, and closed transfer devices move materials between processing zones without opening a barrier to the surrounding room air, minimizing the exposure window during which contamination can occur.
Robotics in API Manufacturing Operations
The global pharmaceutical market is growing rapidly, though estimates vary by scope some analyses put the specialized manufacturing/packaging robotics segment in the low hundreds of millions of dollars growing at 8-13% CAGR through the early 2030s, while broader market definitions that include laboratory and drug-discovery robotics put current market value well into the billions.
What’s consistent across every estimate is the growth rate and the underlying driver regulatory pressure to reduce human-introduced variability. Some industry estimates suggest automation can reduce product defects in pharmaceutical manufacturing by up to 80% relative to comparable manual processes, and that close to 70% of pharmaceutical manufacturers now use some form of robotic technology to reduce contamination and human error risk in production.
Reduced Human Contact During Production Processes
Collaborative robots (cobots) and fully autonomous systems are increasingly deployed for repetitive, high-precision tasks vial filling, powder handling, packaging, and palletizing specifically because reducing manual intervention lowers both contamination risk and batch-to-batch variability at the same time.
Process Automation
Automated process control systems govern parameters like mixing time, temperature ramps, and reaction endpoints with a consistency manual operation cannot replicate, directly supporting both quality and regulatory documentation requirements under 21 CFR Part 11.
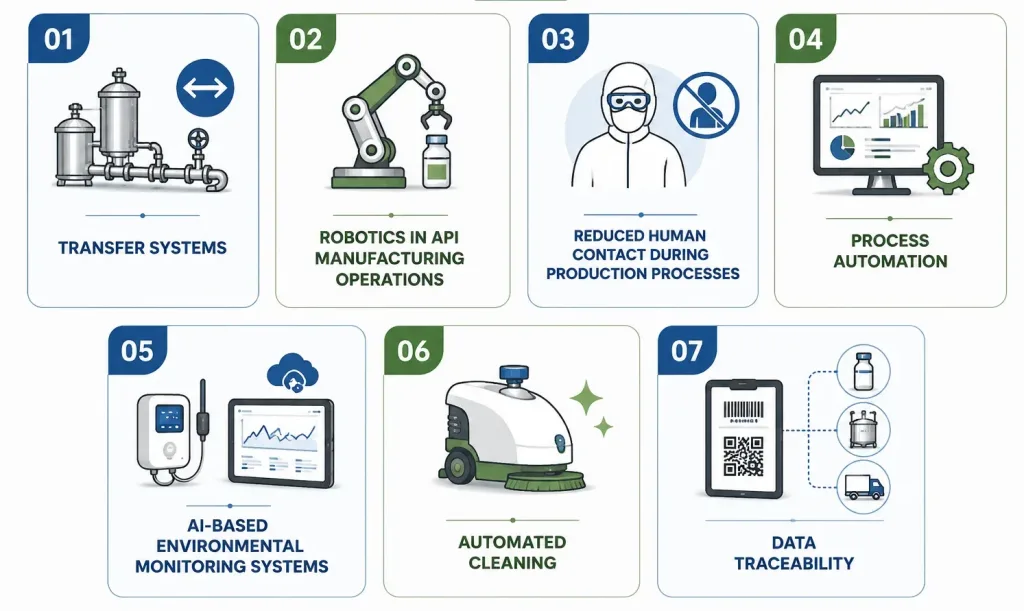
AI-Based Environmental Monitoring Systems
AI-enabled monitoring platforms can identify anomalies in production data in real time, flagging deviations before they escalate into contamination events rather than relying solely on retrospective batch review.
Automated Cleaning
Automated cleaning-in-place (CIP) and sterilization-in-place (SIP) systems, along with automated decontamination cycles in isolators (commonly vaporized hydrogen peroxide, or VHP, systems), remove the variability of manual cleaning execution one of the most frequently cited weak points in FDA inspection findings.
Data Traceability
Automated systems inherently generate the electronic batch records, audit trails, and traceability data that regulators expect closing the loop between physical process control and the documentation that proves it happened correctly.
Cleaning Validation Is a Core Part of Contamination Control
Cleaning validation the documented proof that equipment cleaning procedures reliably remove residues, degradation products, and cleaning agents themselves below defined acceptable limits is a foundational GMP requirement, particularly for multi-product API facilities where cross-contamination between different molecules is a genuine risk.
A validated cleaning process must demonstrate consistent, reproducible results across multiple runs, not a single successful outcome. This typically requires:
For facilities handling multiple APIs on shared equipment, cleaning validation failures are among the most common and most serious findings in regulatory inspections, since they represent a direct cross-contamination pathway from one product to another.
Containment Technologies Are Becoming Increasingly Important
As API portfolios shift toward oncology drugs, hormones, and other high-potency compounds, containment technology has moved from a specialized niche to a mainstream capital investment.
The global pharmaceutical isolator and containment systems market is valued in the range of roughly USD 4-9 billion as of 2025 depending on scope, with most forecasts projecting sustained growth at CAGRs between 6% and 12% through the early 2030s among the faster-growing categories in pharmaceutical manufacturing infrastructure.
Several distinct technology categories fall under this umbrella:
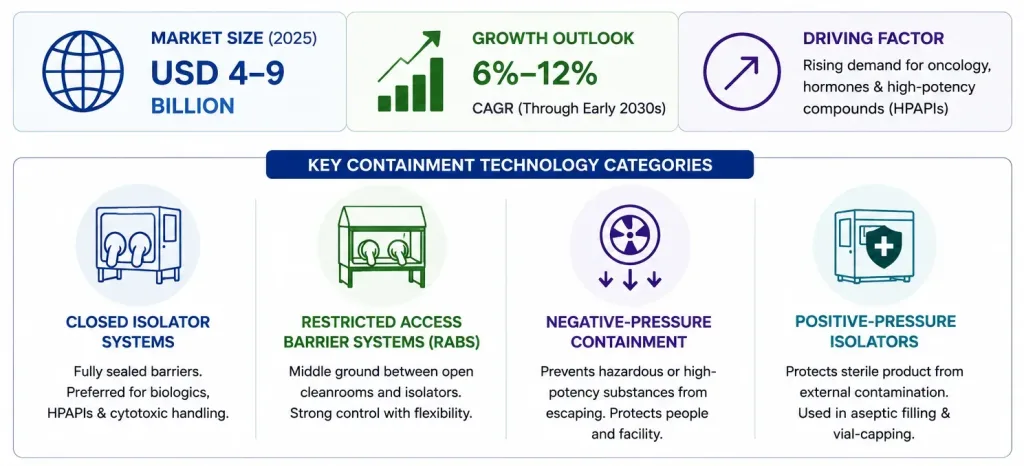
High-Potency API (HPAPI) production specifically is one of the fastest-growing drivers of containment investment, with the broader HPAPI market growing at roughly 7.5-8.8% CAGR faster than the API market overall precisely because these compounds require the specialized handling infrastructure this section describes.
Environmental Monitoring Systems Help Detect Contamination Early
Environmental monitoring (EM) is frequently described as the “eyes and ears” of a contamination control strategy the mechanism by which manufacturers continuously track microbial and particulate contamination levels in cleanrooms and other controlled spaces.
Rather than functioning as a pass/fail checkpoint, effective EM programs use real-time data and trend analysis to detect subtle upward drift in contamination levels before they cross action limits and affect product quality.
Modern EM programs increasingly rely on:
This is precisely the area where regulatory scrutiny is intensifying fastest. Enforcement commentary from GMP compliance specialists in 2025-26 has repeatedly flagged that environmental monitoring data alone without a functioning investigation and CAPA process behind it does not protect a facility from regulatory action; inspectors increasingly probe whether EM excursions were properly investigated and closed out, not merely recorded.
Personnel Training and GMP Culture Are Essential for Risk Prevention
Technology and facility design can only go so far a contamination-free product ultimately starts with people. Industry guidance is explicit that cultivating a genuine “quality culture,” where every employee from lab technicians to senior managers understands and prioritizes contamination control in their daily routines, is a foundational building block of any effective Contamination Control Strategy, not a secondary consideration layered on top of technical controls.
Effective GMP training programs for contamination control typically cover:
Good Manufacturing Practices as a framework exist specifically to ensure that products are consistently produced and controlled according to quality standards, and personnel competence is explicitly one of its core pillars alongside facility design, documentation, and process control.
Raw Material Control Is a Critical Part of Contamination Prevention
Contamination doesn’t only originate on the production floor it frequently enters a facility through incoming raw materials, key starting materials (KSMs), and intermediates. A comprehensive raw material control program addresses several distinct risk points:
Raw Material Supplier Audits
Physical or third-party audits of raw material suppliers verify that upstream handling, storage, and quality systems meet the standard the API manufacturer itself is held to a supplier’s raw material contamination becomes the API manufacturer’s contamination the moment it enters the process.
Quality Verification
Incoming raw materials require independent testing against defined specifications rather than blanket reliance on a supplier’s certificate of analysis, particularly for materials of biological origin where contamination risk is elevated.
Prevention of Cross-Mixing During Material Handling
Dedicated handling equipment, clearly segregated storage, and rigorous material identification protocols prevent one raw material from contaminating another during receipt, sampling, and staging a particular risk in multi-product facilities.
Packaging Integrity
Raw material packaging must be inspected on arrival for damage or tampering, since compromised packaging is a direct contamination entry point long before the material reaches the processing area.
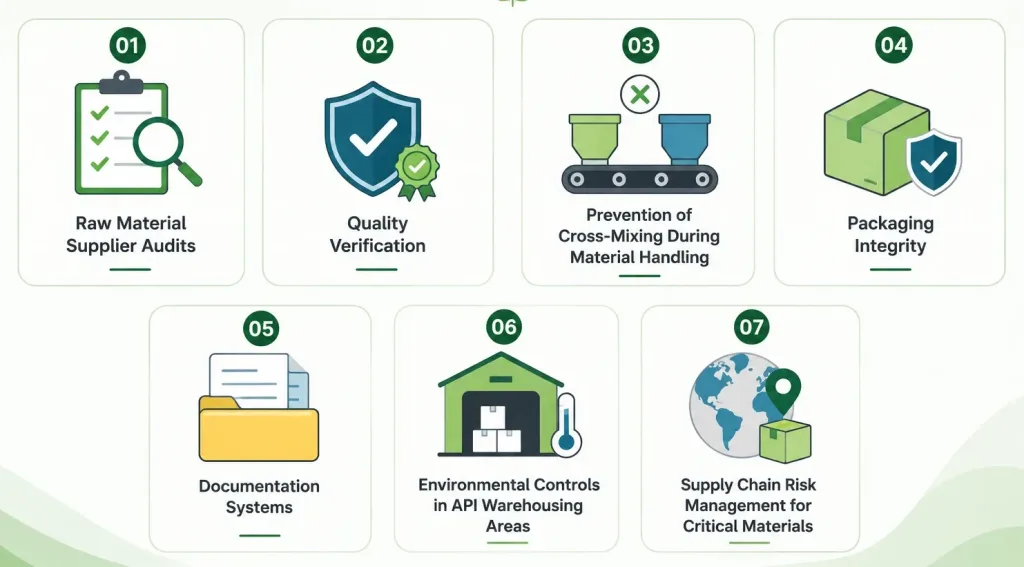
Documentation Systems
Full chain-of-custody documentation from supplier certificate through receipt, sampling, testing, and release is what makes raw material contamination traceable and investigable after the fact, rather than an unexplained anomaly discovered only in the finished product.
Environmental Controls in API Warehousing Areas
Temperature, humidity, and pest control in warehousing directly affect raw material stability and contamination risk before materials ever reach production a frequently underestimated link in the contamination-control chain.
Supply Chain Risk Management for Critical Materials
Given that many APIs still depend on imported KSMs and intermediates, single-source dependency on any one supplier or one geography compounds contamination risk with supply continuity risk a lesson reinforced repeatedly as global buyers have diversified sourcing away from single-country dependency in recent years.
Regulatory Agencies Are Increasingly Strict About Contamination Control
The regulatory data leaves little ambiguity contamination-related enforcement is intensifying, not easing. Analysis of FDA Form 483 observations and subsequent warning letters found that a company receiving a Form 483 has more than a 50% chance of a follow-up warning letter, particularly when the company’s response to the initial observation is inadequate meaning that how a facility handles a contamination-related finding often matters as much as the finding itself.
Several regulatory trends are worth noting:

The clear regulatory signal is that agencies are evaluating not just whether contamination occurred, but whether the underlying quality system investigation rigor, CAPA effectiveness, and data integrity would have caught and corrected it.
Future Technologies Reshaping Contamination Prevention in API Manufacturing
Contamination control is entering a phase defined by digital integration rather than purely physical barriers. Several converging technologies are reshaping how facilities anticipate and prevent contamination rather than simply detecting it after the fact:
Smart Manufacturing Systems
Industry 4.0 principles networked equipment, centralized data platforms, and automated decision-making are being layered onto traditional GMP manufacturing to create facilities that adjust process parameters in real time rather than relying solely on periodic manual checks.
IoT-Based Environmental Monitoring Technologies
Networked sensor arrays feeding continuous data into centralized platforms are becoming a standard feature of new cleanroom builds, enabling facility-wide visibility that legacy point-monitoring systems could not provide.
Advanced Robotics in Cleanroom Operations
Beyond simple task automation, next-generation robotics are being integrated directly into isolators and closed systems, combining containment technology with robotic precision to further reduce the human footprint in critical processing zones.
Digital Twin Technology
Digital twins virtual replicas of physical manufacturing processes allow facilities to simulate the impact of process changes, equipment modifications, or environmental excursions before implementing them physically, reducing the risk that a well-intentioned change introduces a new contamination pathway.
Real-Time Quality Monitoring and Analytics
Rather than releasing batches based on end-point testing alone, real-time analytics increasingly support continued process verification throughout manufacturing, catching deviations as they develop rather than after the batch is complete.
Automated Cleaning
Next-generation automated decontamination including advanced VHP (vaporized hydrogen peroxide) systems integrated directly into isolators continues to reduce reliance on manual cleaning validation cycles.
Data-Driven GMP Compliance
Predictive analytics applied to historical deviation and inspection data are beginning to help facilities identify contamination risk patterns proactively, shifting quality management from reactive investigation toward predictive prevention.
Strategic Checklist for Evaluating Contamination Control Systems
For importers, quality auditors, or partners evaluating whether an API manufacturing facility has a genuinely robust contamination control program rather than one that merely looks compliant on paper the following checklist areas are the ones regulators and experienced auditors consistently probe:
Cleanroom Design Assessment
Verify ISO 14644 classifications is appropriate for the processes performed, confirm HVAC and filtration design documentation, and check whether pressure differentials and airflow patterns match the facility’s actual layout on the ground, not just the design drawings.
Environmental Control Evaluation
Review historical temperature, humidity, and differential pressure trend data not just current readings to assess whether the facility maintains control consistently or only during announced inspections.
Sanitization Process Review
Examine cleaning validation protocols, sanitizing agent rotation programs (to prevent resistant organism buildup), and documented evidence that cleaning procedures are followed exactly as validated, not merely as written.
Containment Systems for High-Potency API Manufacturing
For any facility handling HPAPIs or cytotoxic compounds, confirm the presence of appropriately rated isolators or RABS, verify operator exposure monitoring data, and check containment validation records.

Personnel GMP Training
Ask for training completion records, requalification frequency, and more importantly evidence that training translates into practice through direct observation during the audit itself.
Environmental Monitoring
Review the full EM program sampling locations, frequency, action/alert limits, and critically, how excursions are investigated and closed out, since this is where inspectors increasingly focus.
Raw Material Handling
Trace a sample raw material lot from supplier audit through receipt, testing, storage, and release to confirm the paper trail matches physical practice.
Audit Readiness Evaluation
Assess whether the facility’s documentation, deviation logs, and CAPA records are genuinely audit-ready at any time, or whether they appear to be assembled reactively ahead of scheduled inspections a distinction experienced auditors can usually detect.
Conclusion
The data across regulatory enforcement, market investment, and technology adoption tells a consistent story contamination control has become one of the defining competitive and compliance battlegrounds in API manufacturing. Contamination remains the leading or near-leading cause of drug recalls, regulatory scrutiny is intensifying rather than relaxing, and the industry’s response measured in billions of dollars flowing into cleanrooms, isolators, robotics, and environmental monitoring reflects how seriously manufacturers are being forced to take it.
For any organization evaluating an API manufacturing partner, the message is clear contamination control cannot be assessed from a certificate alone. It requires looking at facility design, automation maturity, cleaning validation rigor, containment technology appropriate to the compounds being handled, environmental monitoring discipline, personnel training culture, and raw material control together, not in isolation. Manufacturers that treat these as an integrated system, continuously monitored and improved rather than periodically inspected, are the ones best positioned to avoid the recalls, warning letters, and import alerts that have become increasingly common across the industry in 2025 and 2026.